

دوره 26، شماره 2 - ( بهار 1399 )
جلد 26 شماره 2 صفحات 191-182 |
برگشت به فهرست نسخه ها



![]()
![]()
![]()
Download citation:
BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:



BibTeX | RIS | EndNote | Medlars | ProCite | Reference Manager | RefWorks
Send citation to:
Shafaat S M, Hashemi M, Majd A, Abiri M, Zeinali S. Genetic Assessment of Mucopolysaccharidosis Type IV and the First Pathological Mutation of c.313A>G in the Iranian Population. Intern Med Today 2020; 26 (2) :182-191
URL: http://imtj.gmu.ac.ir/article-1-3413-fa.html
URL: http://imtj.gmu.ac.ir/article-1-3413-fa.html
شفاعت سید مهدی، هاشمی مهرداد، مجد احمد، عبیری مریم، زینلی سیروس. بررسی ژنتیکی دو بیمار ایرانی مبتلا به موکوپلی ساکاریدوز تیپ 4 (سندرم مورکیو) با استفاده از تکنیک Next Generation Sequencing) NGS) و مشاهده جهش بیماریزای c.313A>G برای اولینبار در جمعیت ایرانی. طب داخلی روز. 1399; 26 (2) :182-191
سید مهدی شفاعت1 
 ، مهرداد هاشمی2 ، احمد مجد1 ، مریم عبیری3 ، سیروس زینلی*
، مهرداد هاشمی2 ، احمد مجد1 ، مریم عبیری3 ، سیروس زینلی*  4
4
، مهرداد هاشمی2 ، احمد مجد1 ، مریم عبیری3 ، سیروس زینلی* 4
1- گروه زیستشناسی، دانشگاه آزاد اسلامی واحد تهران شمال، تهران، ایران.
2- گروه ژنتیک مولکولی، دانشگاه آزاد اسلامی واحد پزشکی تهران، تهران، ایران.
3- گروه ژنتیک و بیولوژی مولکولی، دانشگاه علومپزشکی ایران، تهران، ایران.
4- گروه پزشکی مولکولی، مرکز تحقیقات بیوتکنولوژی، انستیتو پاستور ایران، تهران، ایران. ، zeinali@kawsar.ir
2- گروه ژنتیک مولکولی، دانشگاه آزاد اسلامی واحد پزشکی تهران، تهران، ایران.
3- گروه ژنتیک و بیولوژی مولکولی، دانشگاه علومپزشکی ایران، تهران، ایران.
4- گروه پزشکی مولکولی، مرکز تحقیقات بیوتکنولوژی، انستیتو پاستور ایران، تهران، ایران. ، zeinali@kawsar.ir
متن کامل [PDF 3796 kb]
(1506 دریافت)
| چکیده (HTML) (3407 مشاهده)



References
Bouwman MG, Teunissen QG, Wijburg FA, Linthorst GE. ‘Doctor Google’ ending the diagnostic odyssey in lysosomal storage disorders: Parents using internet search engines as an efficient diagnostic strategy in rare diseases. Archives of Disease in Childhood. 2010; 95(8):642-4. [DOI:10.1136/adc.2009.171827] [PMID]
Wraith JE. The mucopolysaccharidoses: A clinical review and guide to management. Archives of Disease in Childhood. 1995; 72(3):263-7. [DOI:10.1136/adc.72.3.263] [PMID] [PMCID]
Sheth J, Patel P, Sheth F, Shah R. Lysosomal storage disorders. Indian Pediatrics. 2004; 41(3):260-5. [PMID]
Bleier M, Yuskiv N, Priest T, Moisa Popurs MA, Stockler-Ipsiroglu S, BC Children’s Hospital, et al. Morquio B patient/caregiver survey: First insight into the natural course of a rare GLB1 related condition. Molecular Genetics and Metabolism Reports. 2018; 16:57-63. [DOI:10.1016/j.ymgmr.2018.06.006] [PMID] [PMCID]
Rivera-Colón Y, Schutsky EK, Kita AZ, Garman SC. The structure of human GALNS reveals the molecular basis for mucopolysaccharidosis IV A. Journal of Molecular Biology. 2012; 423(5):736-51. [DOI:10.1016/j.jmb.2012.08.020] [PMID] [PMCID]
Hofer D, Paul K, Fantur K, Beck M, Bürger F, Caillaud C, et al. GM1 gangliosidosis and Morquio B disease: Expression analysis of missense mutations affecting the catalytic site of acid beta-galactosidase. Human Mutation. 2009; 30(8):1214-21. [DOI:10.1002/humu.21031] [PMID]
Santamaria R, Chabás A, Coll MJ, Miranda CS, Vilageliu L, Grinberg D. Twenty-one novel mutations in the GLB1 gene identified in a large group of GM1-gangliosidosis and Morquio B patients: Possible common origin for the prevalent p.R59H mutation among gypsies. Human Mutation. 2006; 27(10):1060. [DOI:10.1002/humu.9451] [PMID]
Lei HL, Ye J, Qiu WJ, Zhang HW, Han LS, Wang Y, et al. Beta-galactosidase deficiencies and novel GLB1 mutations in three Chinese patients with Morquio B disease or GM1 gangliosidosis. World Journal of Pediatrics. 2012; 8(4):359-62. [DOI:10.1007/s12519-012-0382-0] [PMID]
Gucev ZS, Tasic V, Jancevska A, Zafirovski G, Kremensky I, Sinigerska I, et al. Novel beta-galactosidase gene mutation p.W273R in a woman with mucopolysaccharidosis type IVB (Morquio B) and lack of response to in vitro chaperone treatment of her skin fibroblasts. American Journal of Medical Genetics. Part A. 2008; 146A(13):1736-40. [DOI:10.1002/ajmg.a.32318] [PMID]
Paschke E, Milos I, Kreimer-Erlacher H, Hoefler G, Beck M, Hoeltzenbein M, et al. Mutation analyses in 17 patients with deficiency in acid beta-galactosidase: Three novel point mutations and high correlation of mutation W273L with Morquio disease type B. Human Genetics. 2001; 109(2):159-66. [DOI:10.1007/s004390100570] [PMID]
Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Research. 1988; 16(3):1215. [DOI:10.1093/nar/16.3.1215] [PMID] [PMCID]
Caciotti A, Garman SC, Rivera-Colón Y, Procopio E, Catarzi S, Ferri L, et al. GM1 gangliosidosis and Morquio B disease: An update on genetic alterations and clinical findings. Biochimica et Biophysica Acta. 2011; 1812(7):782-90. [DOI:10.1016/j.bbadis.2011.03.018] [PMID] [PMCID]
Shafaat M, Alaee MR, Rahmanifar A, Setoodeh A, Razzaghy-Azar M, Bagherian H, et al. Autozygosity mapping of methylmalonic acidemia associated genes by short tandem repeat markers facilitates the identification of five novel mutations in an Iranian patient cohort. Metabolic Brain Disease. 2018; 33(5):1689-97. [DOI:10.1007/s11011-018-0277-4] [PMID]
Shafaat M, Hashemi M, Majd A, Abiri M, Zeinali S. Genetic testing of mucopolysaccharidoses disease using multiplex PCR- based panels of STR markers: In silico analysis of novel mutations. Metabolic Brain Disease. 2019; 34(5):1447-55. [DOI:10.1007/s11011-019-00434-z] [PMID]
Venselaar H, Te Beek TA, Kuipers RK, Hekkelman ML, Vriend G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics. 2010; 11:548. [DOI:10.1186/1471-2105-11-548] [PMID] [PMCID]
متن کامل: (3960 مشاهده)
مقدمه
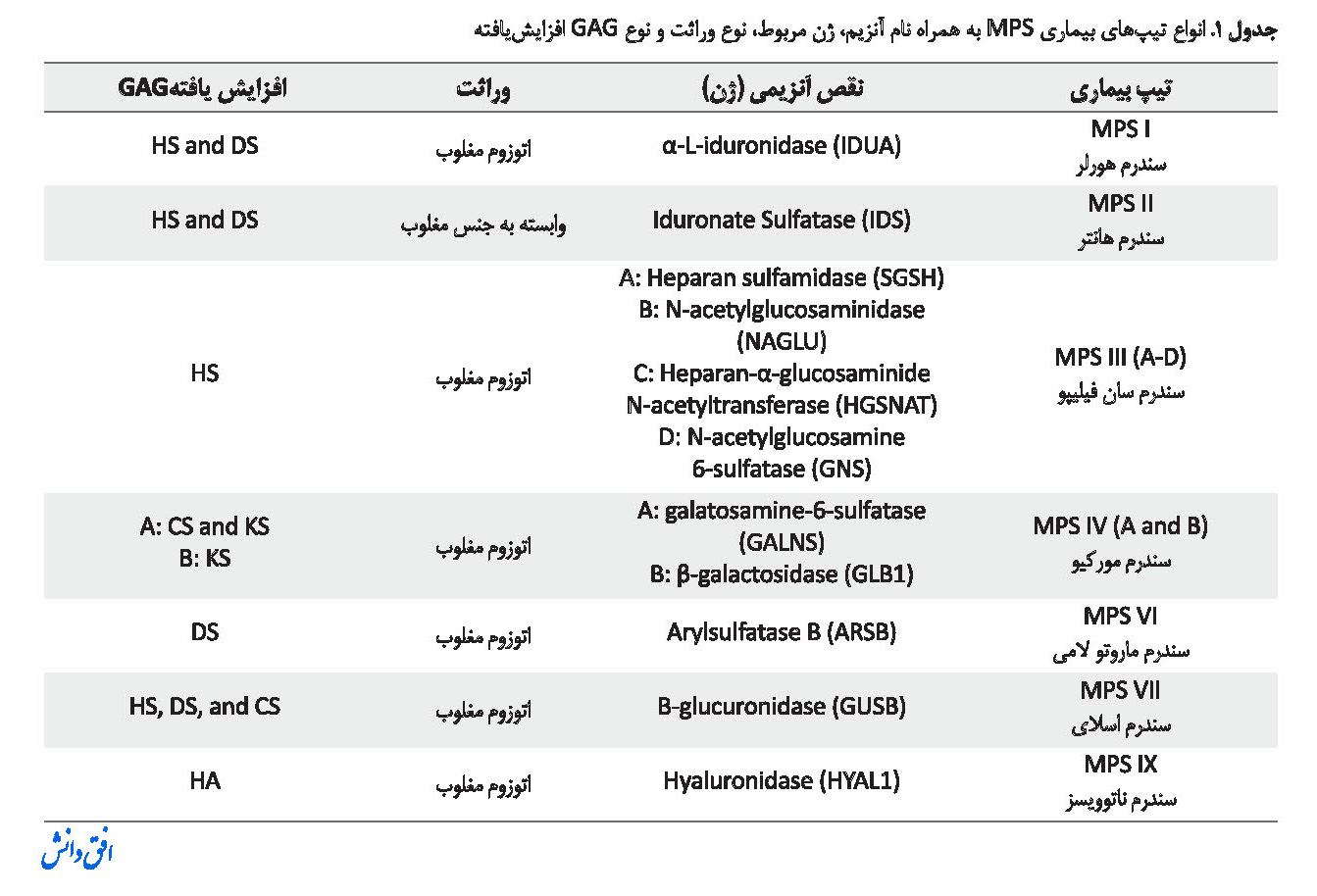
بیماریهای ذخیرهای لیزوزومی (LSD) حدوداً 50 بیماری را شامل میشوند که وجه اشتراک تمامی آنها تجمع یکسری از مواد زاید (ماکرومولکولها) در اندامک لیزوزوم سلولهاست [1]. یکی از معروفترین بیماریهای ذخیرهای لیزوزومی بیماری موکوپلی ساکاریدوزیس (MPS) است که نوعی بیماری متابولیکی وراثتی و ناشی از نقص در گروهی از آنزیمهای لیزوزومی است که نقش این آنزیمها شکستن و تخریب زنجیرههای طویل کربوهیدراتی به نام گلیکوزآمینوگلیکانها (GAGs) است که این پلیساکاریدها ناشی از تکرارهای بسیار زیادی از دیساکاریدها هستند. GAGهایی که سبب این بیماری میشوند شامل هپاران سولفات (HS)، درماتان سولفات (DS)، کراتان سولفات (KS)، کندروایتین سولفات (CS) و هیالورونان هستند [3 ،2]. در بیماری MPS تجمع این مواد در اندامک لیزوزوم در سلولها صورت میگیرد که سپس در جریان خون ترشح میشوند و درنهایت از طریق ادرار دفع میشوند. نقص در 11 آنزیم سبب افزایش این پنج پلیساکارید در بیماران میشود که درمجموع باعث به وجود آمدن هفت تیپ متفاوت بیماری MPS میشود (در جدول شماره 1 نام آنزیمها و نوع بیماری MPS به وجود آمده توضیح داده شده است).
تیپ چهارم بیماری MPS سندرم مورکیو نام دارد که این تیپ بیماری خود به دو دسته تقسیم میشود و دو ژن متفاوت از هم هرکدام میتوانند بهتنهایی باعث بروز این بیماری شوند. وراثت هر دو ژن از نوع اتوزوم مغلوب است و بنابراین این تیپ در ازدواجهای خویشاوندی شایعتر است [4].
شایعترین تیپ MPSIV تیپ A آن است که این تیپ بیماری ناشی از کمبود یا عدم حضور آنزیم گالاکتوزآمین 6 سولفاتاز (EC 3.1.6.4) است. ژن SNLAG (NM_000512.4) مسئول تولید این آنزیم در سلولهاست که این ژن 522 اسیدامینه را کد میکند و دارای 14 اگزون است. جایگاه کروموزومی این ژن 16q24.3 است. آنزیم گالاکتوزآمین 6 سولفاتاز مسئول حذف گروههای سولفات از N ترمینال استیل گالاکتوزآمین 6 سولفات در موکوپلی ساکاریدهایی چون کراتان سولفات و کندروایتین سولفات است. درنتیجه در بیماران MPSIV A به دلیل نقص ژنی در این آنزیم، میزان GAG افزایشیافته در سلولها، کراتان سولفات و کندروایتین سولفات است. شیوع این تیپ بیماری، یک در هر 200 هزار تولد نوزاد زنده در جهان تخمین زده شده است [5]. علائم بالینی مورکیو A شامل اختلالات اسکلتی، کاهش شنوایی، مشکلاتی در قرنیه چشمها، نارساییهای قلبی، کوتاه بودن غیرطبیعی قد، اسکلیوز، کیفوز، نقص مینای دندانها، سفتی بیش از حد مفاصل، پیشانی برجسته و گردن کوتاه است. علائم این تیپ از بیماری معمولاً در دوسالگی با افزایش کراتان سولفات و کندروایتین سولفات بروز پیدا میکند که اولین علائم ظاهرشونده هم اختلالات اسکلتی و انحنای ستون فقرات است [5].
تیپ دوم MPSIV نوع B آن است که این تیپ بر اثر کمبود یا عدم حضور آنزیم بتا گالاکتوزیداز (β-Gal) (E.C.3.2.1.23) ایجاد میشود. این آنزیم توسط ژنی به نام 1BLG (NM_000404.3)کد میشود. این ژن دارای جایگاه کروموزومی 3p22.3 است که 677 اسیدآمینه را نیز کد میکند و 16 اگزون دارد. نقص ژنی GLB1 غیر از B MPSIV سبب ایجاد بیماری دیگری به نام GM1 gangliosidosis نیز میشود. به عبارتی ایجاد موتاسیون در ژن GLB1 باعث نقص در فعالیت آنزیم β-Gal میشود که این آنزیم برای تجزیه فسفولیپید GM1 gangliosidosis و کراتان سولفات در سلولهای بدن کاربرد دارد. درنتیجه در بیماران B MPSIV ،GAG افزایشیافته در بدن، کراتان سولفات است. این تیپ بیماری معمولاً با تغیرات اسکلتی مانند دیسپلازی اسکلتی و کوتاهی قد همراه است.
مبتلایان بیماری را در دوران نوزادی یا بدو تولد نشان نمیدهند و معمولاً در سن دوسالگی به بعد با مشکلات راه رفتن بروز بیماری را نشان میدهند. رفتهرفته علائم دیگر این بیماری همچون مشکلات تنفسی، مشکلات خواب، بیماریهای دریچه قلبی، اختلالات شنوایی و اختلالات قرنیه چشم نیز با افزایش سن بروز مییابند. هوش در این افراد تا حدی نسبت به افراد سالم پایینتر است [10-6].
بیماریهای ذخیرهای لیزوزومی (LSD) حدوداً 50 بیماری را شامل میشوند که وجه اشتراک تمامی آنها تجمع یکسری از مواد زاید (ماکرومولکولها) در اندامک لیزوزوم سلولهاست [1]. یکی از معروفترین بیماریهای ذخیرهای لیزوزومی بیماری موکوپلی ساکاریدوزیس (MPS) است که نوعی بیماری متابولیکی وراثتی و ناشی از نقص در گروهی از آنزیمهای لیزوزومی است که نقش این آنزیمها شکستن و تخریب زنجیرههای طویل کربوهیدراتی به نام گلیکوزآمینوگلیکانها (GAGs) است که این پلیساکاریدها ناشی از تکرارهای بسیار زیادی از دیساکاریدها هستند. GAGهایی که سبب این بیماری میشوند شامل هپاران سولفات (HS)، درماتان سولفات (DS)، کراتان سولفات (KS)، کندروایتین سولفات (CS) و هیالورونان هستند [3 ،2]. در بیماری MPS تجمع این مواد در اندامک لیزوزوم در سلولها صورت میگیرد که سپس در جریان خون ترشح میشوند و درنهایت از طریق ادرار دفع میشوند. نقص در 11 آنزیم سبب افزایش این پنج پلیساکارید در بیماران میشود که درمجموع باعث به وجود آمدن هفت تیپ متفاوت بیماری MPS میشود (در جدول شماره 1 نام آنزیمها و نوع بیماری MPS به وجود آمده توضیح داده شده است).
تیپ چهارم بیماری MPS سندرم مورکیو نام دارد که این تیپ بیماری خود به دو دسته تقسیم میشود و دو ژن متفاوت از هم هرکدام میتوانند بهتنهایی باعث بروز این بیماری شوند. وراثت هر دو ژن از نوع اتوزوم مغلوب است و بنابراین این تیپ در ازدواجهای خویشاوندی شایعتر است [4].
شایعترین تیپ MPSIV تیپ A آن است که این تیپ بیماری ناشی از کمبود یا عدم حضور آنزیم گالاکتوزآمین 6 سولفاتاز (EC 3.1.6.4) است. ژن SNLAG (NM_000512.4) مسئول تولید این آنزیم در سلولهاست که این ژن 522 اسیدامینه را کد میکند و دارای 14 اگزون است. جایگاه کروموزومی این ژن 16q24.3 است. آنزیم گالاکتوزآمین 6 سولفاتاز مسئول حذف گروههای سولفات از N ترمینال استیل گالاکتوزآمین 6 سولفات در موکوپلی ساکاریدهایی چون کراتان سولفات و کندروایتین سولفات است. درنتیجه در بیماران MPSIV A به دلیل نقص ژنی در این آنزیم، میزان GAG افزایشیافته در سلولها، کراتان سولفات و کندروایتین سولفات است. شیوع این تیپ بیماری، یک در هر 200 هزار تولد نوزاد زنده در جهان تخمین زده شده است [5]. علائم بالینی مورکیو A شامل اختلالات اسکلتی، کاهش شنوایی، مشکلاتی در قرنیه چشمها، نارساییهای قلبی، کوتاه بودن غیرطبیعی قد، اسکلیوز، کیفوز، نقص مینای دندانها، سفتی بیش از حد مفاصل، پیشانی برجسته و گردن کوتاه است. علائم این تیپ از بیماری معمولاً در دوسالگی با افزایش کراتان سولفات و کندروایتین سولفات بروز پیدا میکند که اولین علائم ظاهرشونده هم اختلالات اسکلتی و انحنای ستون فقرات است [5].
تیپ دوم MPSIV نوع B آن است که این تیپ بر اثر کمبود یا عدم حضور آنزیم بتا گالاکتوزیداز (β-Gal) (E.C.3.2.1.23) ایجاد میشود. این آنزیم توسط ژنی به نام 1BLG (NM_000404.3)کد میشود. این ژن دارای جایگاه کروموزومی 3p22.3 است که 677 اسیدآمینه را نیز کد میکند و 16 اگزون دارد. نقص ژنی GLB1 غیر از B MPSIV سبب ایجاد بیماری دیگری به نام GM1 gangliosidosis نیز میشود. به عبارتی ایجاد موتاسیون در ژن GLB1 باعث نقص در فعالیت آنزیم β-Gal میشود که این آنزیم برای تجزیه فسفولیپید GM1 gangliosidosis و کراتان سولفات در سلولهای بدن کاربرد دارد. درنتیجه در بیماران B MPSIV ،GAG افزایشیافته در بدن، کراتان سولفات است. این تیپ بیماری معمولاً با تغیرات اسکلتی مانند دیسپلازی اسکلتی و کوتاهی قد همراه است.
مبتلایان بیماری را در دوران نوزادی یا بدو تولد نشان نمیدهند و معمولاً در سن دوسالگی به بعد با مشکلات راه رفتن بروز بیماری را نشان میدهند. رفتهرفته علائم دیگر این بیماری همچون مشکلات تنفسی، مشکلات خواب، بیماریهای دریچه قلبی، اختلالات شنوایی و اختلالات قرنیه چشم نیز با افزایش سن بروز مییابند. هوش در این افراد تا حدی نسبت به افراد سالم پایینتر است [10-6].
در این طرح تحقیقاتی دو بیمار مبتلا به MPSIV مورد بررسی ژنتیکی قرار گرفتند تا جهشهای عامل بروز این بیماری در آنها مشخص شود. هر دو مبتلا از طریق تکنیک SGN توالی یابی شدند تا درنهایت جهش ژنتیکی عامل بروز بیماری در آن ها مشاهده شد.
مواد و روشها
جمعآوری نمونه
در این مطالعه دو کودک ایرانی مبتلا به سندرم مورکیو که هر دو حاصل ازدواج خویشاوندی بودند، به آزمایشگاه ژنتیک پزشکی دکتر زینلی معرفی شدند. معیارهای ورود این بیماران در این مطالعه پایین بودن سطح آنزیمهای دخیل در این تیپ بیماری MPS از طریق آزمایشات آنزیمی و تشخیص پزشکان متخصص بود. از هر دو خانواده رضایتنامه کتبی مبنی بر انجام تحقیق روی نمونه فرد مبتلا و خانواده او دریافت شد.
مبتلای اول پسری سهساله بود که پدر و مادر او با یکدیگر دخترعمو پسرعمو بودند. اولین علائم ناهنجاری در این کودک تقریبا در 18ماهگی و عفونت ریوی شدید و تشنج و سر بزرگتر از حد طبیعی بوده است. سپس با افزایش سن، علائم بالینی دیگری که شامل لاغری بیش از حد پاها، عدم رشد دندانها، لثه اضافی، چهره کاملاً متورم و خشن، سر بزرگتر از حد طبیعی، عدم تکلم، از دست دادن کامل بینایی، کند ذهنی و نشان ندادن هیچ پاسخی به سر و صدا، سرفههای بیش از حد و مصرف داروهای ریوی، قادر نبودن به نشستن و ایستادن، کیفوز، بیرونزدگی جناغ سینه، عدم تکان دادن انگشتان دست، هپاتومگالی و اسپلنومگالی هستند، در او دیده شده است.
مواد و روشها
جمعآوری نمونه
در این مطالعه دو کودک ایرانی مبتلا به سندرم مورکیو که هر دو حاصل ازدواج خویشاوندی بودند، به آزمایشگاه ژنتیک پزشکی دکتر زینلی معرفی شدند. معیارهای ورود این بیماران در این مطالعه پایین بودن سطح آنزیمهای دخیل در این تیپ بیماری MPS از طریق آزمایشات آنزیمی و تشخیص پزشکان متخصص بود. از هر دو خانواده رضایتنامه کتبی مبنی بر انجام تحقیق روی نمونه فرد مبتلا و خانواده او دریافت شد.
مبتلای اول پسری سهساله بود که پدر و مادر او با یکدیگر دخترعمو پسرعمو بودند. اولین علائم ناهنجاری در این کودک تقریبا در 18ماهگی و عفونت ریوی شدید و تشنج و سر بزرگتر از حد طبیعی بوده است. سپس با افزایش سن، علائم بالینی دیگری که شامل لاغری بیش از حد پاها، عدم رشد دندانها، لثه اضافی، چهره کاملاً متورم و خشن، سر بزرگتر از حد طبیعی، عدم تکلم، از دست دادن کامل بینایی، کند ذهنی و نشان ندادن هیچ پاسخی به سر و صدا، سرفههای بیش از حد و مصرف داروهای ریوی، قادر نبودن به نشستن و ایستادن، کیفوز، بیرونزدگی جناغ سینه، عدم تکان دادن انگشتان دست، هپاتومگالی و اسپلنومگالی هستند، در او دیده شده است.
مبتلای دوم پسری 14ساله است که او نیز حاصل ازدواج خویشاوندی است و در 11سالگی بیماری را با عدم تعادل در راه رفتن نشان داده است و در حال حاضر عدم رشد کافی قد، جلو بودن بیش از حد فک، بینایی ضعیف، جلو بودن غیرطبیعی جناغ سینه و درد لگن و مفاصل زانو نیز در او تظاهر یافتهاند.
مراحل انجام کار
پس از انجام مشاوره ژنتیک، از بیمار و تمام اعضای خانواده ایشان سیسیب10 خون وریدی دریافت شد. سپس DNA ژنومی به روش نمک اشباع [11] تخلیص شد. DNA تخلیصشده هر دو مبتلا NGS شد تا جهشهای عامل بیماری در دو مبتلا با سرعت بیشتری پیدا شود. پس از دریافت نتایج تعیین توالی، در مبتلای یک در ژن GLB1 و در مبتلای دو در ژن GALNS جهش عامل بیماری به صورت هموزیگوت مشاهده شد. سپس اگزونی که در هر مبتلا به عنوان جهش عامل بیماری دیده شده بود، با استفاده از تکنیک تعیین توالی سنگر تعیین توالی شد تا جهش مشاهدهشده در دو مبتلا تأیید شود. سپس هر دو جهش پیدا شده از لحاظ بیماریزایی مورد بررسی قرار گرفتند تا صحت بیماریزا بودن آنها تأیید شود.
یافتهها
نتایج حاصل از NGS به این صورت بود که مبتلای شماره یک دارای جهش c.443G>A در ژن GLB1 به صورت هموزیگوت در اگزون چهار بود که این جهش پیش از این مطالعه در جمعیت دیگری نیز گزارش شده بود و بیماریزایی آن تأیید شده بود [12]. مبتلای شماره دو نیز دارای جهش c.313A>G در ژن GALNS به شکل هموزیگوت و در اگزون سه این ژن بود که این جهش برای اولینبار در این مطالعه دیده شد و تا به امروز گزارش نشده بود (نتایج گزارششده NGS در تصویر شماره 1 نشان داده شده است). سپس برای تأیید این جهشها، اگزون چهار ژن GLB1 در مبتلای یک و اگزون سه ژن GALNS در مبتلای دو با روش سنگر تعیین توالی شد که در هر دو مبتلا جهشهای پیداشده با روش سنگر نیز تأیید شدند (نتایج تعیین توالی سنگر این اگزونها نیز در تصویر شماره 2 نشان داده شده است).
پس از انجام مشاوره ژنتیک، از بیمار و تمام اعضای خانواده ایشان سیسیب10 خون وریدی دریافت شد. سپس DNA ژنومی به روش نمک اشباع [11] تخلیص شد. DNA تخلیصشده هر دو مبتلا NGS شد تا جهشهای عامل بیماری در دو مبتلا با سرعت بیشتری پیدا شود. پس از دریافت نتایج تعیین توالی، در مبتلای یک در ژن GLB1 و در مبتلای دو در ژن GALNS جهش عامل بیماری به صورت هموزیگوت مشاهده شد. سپس اگزونی که در هر مبتلا به عنوان جهش عامل بیماری دیده شده بود، با استفاده از تکنیک تعیین توالی سنگر تعیین توالی شد تا جهش مشاهدهشده در دو مبتلا تأیید شود. سپس هر دو جهش پیدا شده از لحاظ بیماریزایی مورد بررسی قرار گرفتند تا صحت بیماریزا بودن آنها تأیید شود.
یافتهها
نتایج حاصل از NGS به این صورت بود که مبتلای شماره یک دارای جهش c.443G>A در ژن GLB1 به صورت هموزیگوت در اگزون چهار بود که این جهش پیش از این مطالعه در جمعیت دیگری نیز گزارش شده بود و بیماریزایی آن تأیید شده بود [12]. مبتلای شماره دو نیز دارای جهش c.313A>G در ژن GALNS به شکل هموزیگوت و در اگزون سه این ژن بود که این جهش برای اولینبار در این مطالعه دیده شد و تا به امروز گزارش نشده بود (نتایج گزارششده NGS در تصویر شماره 1 نشان داده شده است). سپس برای تأیید این جهشها، اگزون چهار ژن GLB1 در مبتلای یک و اگزون سه ژن GALNS در مبتلای دو با روش سنگر تعیین توالی شد که در هر دو مبتلا جهشهای پیداشده با روش سنگر نیز تأیید شدند (نتایج تعیین توالی سنگر این اگزونها نیز در تصویر شماره 2 نشان داده شده است).
بحث
سندرم مورکیو تیپ چهارم بیماری MPS است که دارای دو تیپ A و B است. شیوع ابتلا به این بیماری در کشورهای خاورمیانه همانند ایران به نسبت دیگر نقاط جهان به علت نرخ بالای ازدواج خویشاوندی بیشتر باشد (نرخ ازدواج خویشاوندی در ایران حدودا 38/6 درصد است [14 ،13]. در این مطالعه دو بیمار که یکی مبتلا به مورکیو A و دیگری مورکیو B بودند مورد بررسی ژنتیکی قرار گرفتند تا جهش عامل بیماری آنها در ژنهای GLB1 و GALNS پیدا شود.
در این مطالعه جهش c.313A>G در ژن GALNS برای اولینبار در یکی از مبتلایان مشاهده شد که تا به حال در دنیا گزارش نشده است. این جهش سبب جابهجا شدن اسیدآمینه آرژنین با گلاسین در کدون 105 میشود. طبق نتیجه NGS، این جهش از نوع Class 2 و بیماریزاست. آرژنین یک اسیدآمینه باردار مثبت و هیدروفیل است، اما گلایسین خنثی و هیدروفوب است و کاملاً با یکدیگر تفاوت دارند. همچنین گلایسین به لحاظ ساختاری کوچکتر از آرژنین است (تصویر شماره 3 الف). در فرم طبیعی پروتئین آرژنین در جایگاه خود یک پل نمکی با گلوتامیک اسید در جایگاه 410 تشکیل میدهد که این موتاسیون کاملاً این میانکنش را به دلیل از بین رفتن بار یونی مناسب برهم میزند. آرژنین به دلیل بار مثبتش با گلوتامیک اسید که دارای بار منفی است در این میانکنش حضور دارد. همچنین این ناحیه به عنوان یک دومین کاتالیتیکی عمل میکند که این موتاسیون کاملاً این نقش را از بین میبرد. گلایسین از لحاظ ساختاری اسیدآمینهای انعطافپذیر است که این ناحیه از پروتئین باید سخت و محکم باشد و وجود گلایسین در این ناحیه کاملاً ساختار پروتئین را تغییر داده و در عملکرد آن، تأثیر مستقیم میگذارد (تصویر شماره 3 ب) [15].
جهش c.443G>A در ژن GLB1 جهش دیگری بود که در این مطالعه در یک مبتلا به صورت هموزیگوت دیده شد و پیش از این مطالعه در دنیا به عنوان یک جهش بیماریزا گزارش شده بود. کاسیوتی و همکارانش در سال 2011، 21 بیمار MPS را مورد آنالیز ژنتیکی قرار دادند که چهار بیمار آنها نوع سندرم مورکیو B بودند. در نتیجه تعیین توالی ژن GLB1 در این چهار بیمار موتاسیون c.443G>A در یکی از بیماران برای اولینبار مشاهده شد. [12]. این جهش سبب جابهجا شدن اسیدآمینه آرژنین با هیستیدین در کدون 148 ژن GLB1 میشود. به لحاظ اندازه، آرژنین بزرگتر از هیستیدین است. در فرم طبیعی، پروتئین آرژنین در جایگاه خود با گلوتامیک اسید در جایگاه 186 پیوند هیدروژنی میدهد. تفاوت اندازه آرژنین با هیستیدین میتواند در ایجاد این پیوند هیدروژنی با گلوتامیک اسید تداخل ایجاد کند و مانع تشکیل این پیوند شود. همچنین اسیدآمینه آرژنین در این موقعیت با گلوتامیک اسید در موقعیت 186، گلوتامیک اسید در موقعیت 188 و آسپارتیک اسید در موقعیت 221 پل نمکی تشکیل میدهد که تبدیل شدن این اسیدآمینه به هیستیدین در تشکیل این پلهای نمکی نیز مشکلساز میشود [15]. درنتیجه این جهش نیز به عنوان یک جهش بیماریزا در ژن GLB1 تلقی میشود.
برای یافتن جهشهای بیماریزای بیشتر در بیماری MPSIV لازم است نمونههای بیشتری از تمامی مناطق ایران تهیه شود تا بتوان جهشهای ژنتیکی بیشتر و گزارشنشده در دنیا را پیدا کرد. از این طریق میتوان به همبستگی ژنوتیپ فنوتیپ در این بیماری نیز دست یافت. با بررسی تعداد نمونههای مبتلای بیشتر میتوان اگزونهای شایع ژنهای GALNS و GLB1 را از نظر میزان جهشپذیری نیز مورد بررسی قرار داد.
بحث و نتیجهگیری
با توجه به مطالعات انجامشده در دنیا در ارتباط با انواع تیپهای بیماری موکوپلی ساکاریدوز، امیدواریم نتایج این مطالعه کمک بزرگی به تشخیص پیش از تولد بیماری موکوپلی ساکاریدوز تیپ چهار بکند. با توجه به اینکه در این مطالعه یک جهش جدید و گزارشنشده در دنیا برای اولینبار دیده شد، پیشنهاد میشود که جامعه آماری بزرگتری از بیماری موکوپلی ساکاریدوز مورد بررسی ژنتیکی قرار بگیرند تا بتوان جهشهای بیماریزای بیشتری را در مبتلایان ایرانی گزارش داد.
ملاحظات اخلاقی
پیروی از اصول اخلاق پژوهش
پیروی از اصول اخلاق پژوهش مطالعه حاضر در کمیته اخلاق مرکز پژوهشی ژنتیک انسانی کوثر به تأیید رسیده است و شماره فرم پیوست انجام آن 6299/98 است.
حامی مالی
این مطالعه از پایاننامه دکترای سید مهدی شفاعت در رشته زیست شناسی گرایش ژنتیک مولکولی دانشگاه آزاد اسلامی واحد تهران شمال استخراج شده است.
مشارکت نویسندگان
تمام نویسندگان در طراحی، اجرا و نگارش همه بخشهای پژوهش حاضر مشارکت داشتهاند.
تعارض منافع
بنابر اظهار نویسندگان، این مقاله تعارض منافع ندارد.
تشکر و قدردانی
از تمام همکاران عزیز آزمایشگاه ژنتیک پزشکی دکتر زینلی و دانشگاه آزاد واحد تهران شمال که در نوشتن این مقاله کمک بزرگی کردند، تشکر و قدردانی میشود.
سندرم مورکیو تیپ چهارم بیماری MPS است که دارای دو تیپ A و B است. شیوع ابتلا به این بیماری در کشورهای خاورمیانه همانند ایران به نسبت دیگر نقاط جهان به علت نرخ بالای ازدواج خویشاوندی بیشتر باشد (نرخ ازدواج خویشاوندی در ایران حدودا 38/6 درصد است [14 ،13]. در این مطالعه دو بیمار که یکی مبتلا به مورکیو A و دیگری مورکیو B بودند مورد بررسی ژنتیکی قرار گرفتند تا جهش عامل بیماری آنها در ژنهای GLB1 و GALNS پیدا شود.
در این مطالعه جهش c.313A>G در ژن GALNS برای اولینبار در یکی از مبتلایان مشاهده شد که تا به حال در دنیا گزارش نشده است. این جهش سبب جابهجا شدن اسیدآمینه آرژنین با گلاسین در کدون 105 میشود. طبق نتیجه NGS، این جهش از نوع Class 2 و بیماریزاست. آرژنین یک اسیدآمینه باردار مثبت و هیدروفیل است، اما گلایسین خنثی و هیدروفوب است و کاملاً با یکدیگر تفاوت دارند. همچنین گلایسین به لحاظ ساختاری کوچکتر از آرژنین است (تصویر شماره 3 الف). در فرم طبیعی پروتئین آرژنین در جایگاه خود یک پل نمکی با گلوتامیک اسید در جایگاه 410 تشکیل میدهد که این موتاسیون کاملاً این میانکنش را به دلیل از بین رفتن بار یونی مناسب برهم میزند. آرژنین به دلیل بار مثبتش با گلوتامیک اسید که دارای بار منفی است در این میانکنش حضور دارد. همچنین این ناحیه به عنوان یک دومین کاتالیتیکی عمل میکند که این موتاسیون کاملاً این نقش را از بین میبرد. گلایسین از لحاظ ساختاری اسیدآمینهای انعطافپذیر است که این ناحیه از پروتئین باید سخت و محکم باشد و وجود گلایسین در این ناحیه کاملاً ساختار پروتئین را تغییر داده و در عملکرد آن، تأثیر مستقیم میگذارد (تصویر شماره 3 ب) [15].
جهش c.443G>A در ژن GLB1 جهش دیگری بود که در این مطالعه در یک مبتلا به صورت هموزیگوت دیده شد و پیش از این مطالعه در دنیا به عنوان یک جهش بیماریزا گزارش شده بود. کاسیوتی و همکارانش در سال 2011، 21 بیمار MPS را مورد آنالیز ژنتیکی قرار دادند که چهار بیمار آنها نوع سندرم مورکیو B بودند. در نتیجه تعیین توالی ژن GLB1 در این چهار بیمار موتاسیون c.443G>A در یکی از بیماران برای اولینبار مشاهده شد. [12]. این جهش سبب جابهجا شدن اسیدآمینه آرژنین با هیستیدین در کدون 148 ژن GLB1 میشود. به لحاظ اندازه، آرژنین بزرگتر از هیستیدین است. در فرم طبیعی، پروتئین آرژنین در جایگاه خود با گلوتامیک اسید در جایگاه 186 پیوند هیدروژنی میدهد. تفاوت اندازه آرژنین با هیستیدین میتواند در ایجاد این پیوند هیدروژنی با گلوتامیک اسید تداخل ایجاد کند و مانع تشکیل این پیوند شود. همچنین اسیدآمینه آرژنین در این موقعیت با گلوتامیک اسید در موقعیت 186، گلوتامیک اسید در موقعیت 188 و آسپارتیک اسید در موقعیت 221 پل نمکی تشکیل میدهد که تبدیل شدن این اسیدآمینه به هیستیدین در تشکیل این پلهای نمکی نیز مشکلساز میشود [15]. درنتیجه این جهش نیز به عنوان یک جهش بیماریزا در ژن GLB1 تلقی میشود.
برای یافتن جهشهای بیماریزای بیشتر در بیماری MPSIV لازم است نمونههای بیشتری از تمامی مناطق ایران تهیه شود تا بتوان جهشهای ژنتیکی بیشتر و گزارشنشده در دنیا را پیدا کرد. از این طریق میتوان به همبستگی ژنوتیپ فنوتیپ در این بیماری نیز دست یافت. با بررسی تعداد نمونههای مبتلای بیشتر میتوان اگزونهای شایع ژنهای GALNS و GLB1 را از نظر میزان جهشپذیری نیز مورد بررسی قرار داد.
بحث و نتیجهگیری
با توجه به مطالعات انجامشده در دنیا در ارتباط با انواع تیپهای بیماری موکوپلی ساکاریدوز، امیدواریم نتایج این مطالعه کمک بزرگی به تشخیص پیش از تولد بیماری موکوپلی ساکاریدوز تیپ چهار بکند. با توجه به اینکه در این مطالعه یک جهش جدید و گزارشنشده در دنیا برای اولینبار دیده شد، پیشنهاد میشود که جامعه آماری بزرگتری از بیماری موکوپلی ساکاریدوز مورد بررسی ژنتیکی قرار بگیرند تا بتوان جهشهای بیماریزای بیشتری را در مبتلایان ایرانی گزارش داد.
ملاحظات اخلاقی
پیروی از اصول اخلاق پژوهش
پیروی از اصول اخلاق پژوهش مطالعه حاضر در کمیته اخلاق مرکز پژوهشی ژنتیک انسانی کوثر به تأیید رسیده است و شماره فرم پیوست انجام آن 6299/98 است.
حامی مالی
این مطالعه از پایاننامه دکترای سید مهدی شفاعت در رشته زیست شناسی گرایش ژنتیک مولکولی دانشگاه آزاد اسلامی واحد تهران شمال استخراج شده است.
مشارکت نویسندگان
تمام نویسندگان در طراحی، اجرا و نگارش همه بخشهای پژوهش حاضر مشارکت داشتهاند.
تعارض منافع
بنابر اظهار نویسندگان، این مقاله تعارض منافع ندارد.
تشکر و قدردانی
از تمام همکاران عزیز آزمایشگاه ژنتیک پزشکی دکتر زینلی و دانشگاه آزاد واحد تهران شمال که در نوشتن این مقاله کمک بزرگی کردند، تشکر و قدردانی میشود.
References
Bouwman MG, Teunissen QG, Wijburg FA, Linthorst GE. ‘Doctor Google’ ending the diagnostic odyssey in lysosomal storage disorders: Parents using internet search engines as an efficient diagnostic strategy in rare diseases. Archives of Disease in Childhood. 2010; 95(8):642-4. [DOI:10.1136/adc.2009.171827] [PMID]
Wraith JE. The mucopolysaccharidoses: A clinical review and guide to management. Archives of Disease in Childhood. 1995; 72(3):263-7. [DOI:10.1136/adc.72.3.263] [PMID] [PMCID]
Sheth J, Patel P, Sheth F, Shah R. Lysosomal storage disorders. Indian Pediatrics. 2004; 41(3):260-5. [PMID]
Bleier M, Yuskiv N, Priest T, Moisa Popurs MA, Stockler-Ipsiroglu S, BC Children’s Hospital, et al. Morquio B patient/caregiver survey: First insight into the natural course of a rare GLB1 related condition. Molecular Genetics and Metabolism Reports. 2018; 16:57-63. [DOI:10.1016/j.ymgmr.2018.06.006] [PMID] [PMCID]
Rivera-Colón Y, Schutsky EK, Kita AZ, Garman SC. The structure of human GALNS reveals the molecular basis for mucopolysaccharidosis IV A. Journal of Molecular Biology. 2012; 423(5):736-51. [DOI:10.1016/j.jmb.2012.08.020] [PMID] [PMCID]
Hofer D, Paul K, Fantur K, Beck M, Bürger F, Caillaud C, et al. GM1 gangliosidosis and Morquio B disease: Expression analysis of missense mutations affecting the catalytic site of acid beta-galactosidase. Human Mutation. 2009; 30(8):1214-21. [DOI:10.1002/humu.21031] [PMID]
Santamaria R, Chabás A, Coll MJ, Miranda CS, Vilageliu L, Grinberg D. Twenty-one novel mutations in the GLB1 gene identified in a large group of GM1-gangliosidosis and Morquio B patients: Possible common origin for the prevalent p.R59H mutation among gypsies. Human Mutation. 2006; 27(10):1060. [DOI:10.1002/humu.9451] [PMID]
Lei HL, Ye J, Qiu WJ, Zhang HW, Han LS, Wang Y, et al. Beta-galactosidase deficiencies and novel GLB1 mutations in three Chinese patients with Morquio B disease or GM1 gangliosidosis. World Journal of Pediatrics. 2012; 8(4):359-62. [DOI:10.1007/s12519-012-0382-0] [PMID]
Gucev ZS, Tasic V, Jancevska A, Zafirovski G, Kremensky I, Sinigerska I, et al. Novel beta-galactosidase gene mutation p.W273R in a woman with mucopolysaccharidosis type IVB (Morquio B) and lack of response to in vitro chaperone treatment of her skin fibroblasts. American Journal of Medical Genetics. Part A. 2008; 146A(13):1736-40. [DOI:10.1002/ajmg.a.32318] [PMID]
Paschke E, Milos I, Kreimer-Erlacher H, Hoefler G, Beck M, Hoeltzenbein M, et al. Mutation analyses in 17 patients with deficiency in acid beta-galactosidase: Three novel point mutations and high correlation of mutation W273L with Morquio disease type B. Human Genetics. 2001; 109(2):159-66. [DOI:10.1007/s004390100570] [PMID]
Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Research. 1988; 16(3):1215. [DOI:10.1093/nar/16.3.1215] [PMID] [PMCID]
Caciotti A, Garman SC, Rivera-Colón Y, Procopio E, Catarzi S, Ferri L, et al. GM1 gangliosidosis and Morquio B disease: An update on genetic alterations and clinical findings. Biochimica et Biophysica Acta. 2011; 1812(7):782-90. [DOI:10.1016/j.bbadis.2011.03.018] [PMID] [PMCID]
Shafaat M, Alaee MR, Rahmanifar A, Setoodeh A, Razzaghy-Azar M, Bagherian H, et al. Autozygosity mapping of methylmalonic acidemia associated genes by short tandem repeat markers facilitates the identification of five novel mutations in an Iranian patient cohort. Metabolic Brain Disease. 2018; 33(5):1689-97. [DOI:10.1007/s11011-018-0277-4] [PMID]
Shafaat M, Hashemi M, Majd A, Abiri M, Zeinali S. Genetic testing of mucopolysaccharidoses disease using multiplex PCR- based panels of STR markers: In silico analysis of novel mutations. Metabolic Brain Disease. 2019; 34(5):1447-55. [DOI:10.1007/s11011-019-00434-z] [PMID]
Venselaar H, Te Beek TA, Kuipers RK, Hekkelman ML, Vriend G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics. 2010; 11:548. [DOI:10.1186/1471-2105-11-548] [PMID] [PMCID]
نوع مطالعه: پژوهشی |
موضوع مقاله:
علوم پايه پزشكي
دریافت: 1398/7/28 | پذیرش: 1398/9/6 | انتشار: 1399/4/1
دریافت: 1398/7/28 | پذیرش: 1398/9/6 | انتشار: 1399/4/1
| بازنشر اطلاعات | |
 |
این مقاله تحت شرایط Creative Commons Attribution-NonCommercial 4.0 International License قابل بازنشر است. |